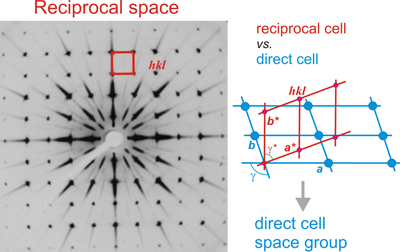
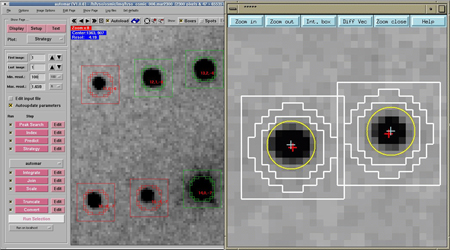
A photographic film showing a reciprocal
plane containing the reciprocal points of type hk0.
Several possible planes of symmetry, marked with the letter m,
are observed.
In short,
the end of a full evaluation of the diffraction
pattern of a crystal means having obtained a complete description of
its reciprocal lattice (geometry + intensities), and hence the
knowledge of the direct lattice: unit
cell constants (a,
b,
c,
α,
β, γ),
lattice
type (primitive or
centered) and
crystal
symmetry (space group), ie, all ingredients to
address the
resolution of the internal structure of the crystal.
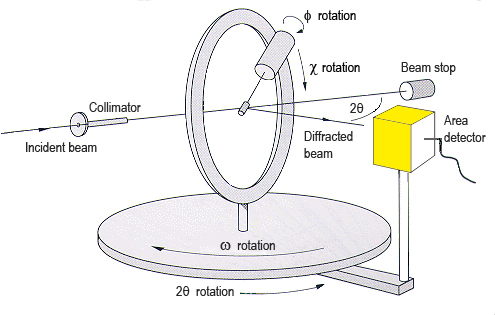
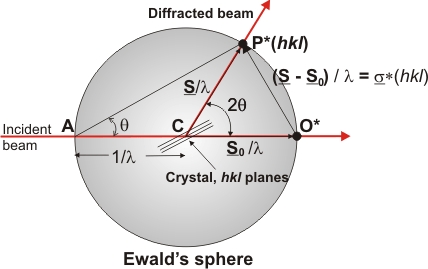
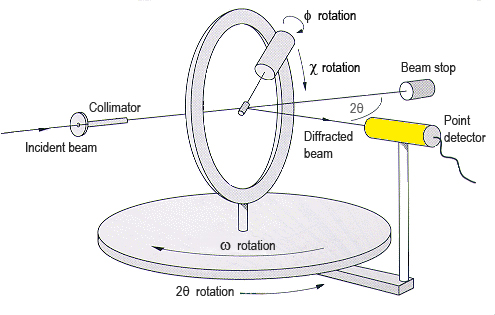
In general, what has been presented up to this point is enough to
understand what the experimental procedures to evaluate the diffraction
pattern are (considering that the diffraction pattern contains Bragg
peaks only). Therefore, the reader could now go
back to the starting
point.
However, the advanced reader might take a look below...
How
many crystals are needeed and at what temperature is the diffraction
experiment done?




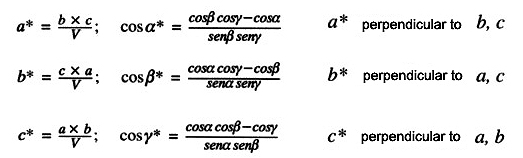



")
")