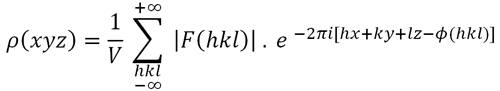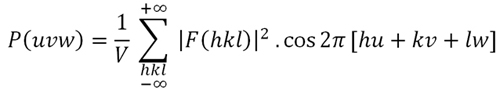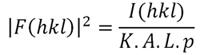- The symmetry of the
Patterson Function (the symmetry of Patterson space) is higher than the
one of the electron
density function (the crystal), so that if the crystal
symmetry can be represented by one of the 230 space groups,
the
corresponding Patterson symmetry will be represented by only 24 space
groups. This simplification is due to the loss of information that
occurs when the structure factors (amplitudes and phases) in ρ(xyz) are
replaced in the P(uvw) function by
the squared amplitudes only. Moreover, the fact that since,
for
instance, there is a vector from atom 1 to atom 2, there will
be
another (identical but in the opposite direction) from atom 2 to atom
1. This means that the Patterson Function is always centrosymmetric
(see figure above). Thus, any maximum of coordinates <u,
v,
w> will always have an equivalent one at <-u,
-v,
-w>.
Therefore, the symmetry of the Patterson Function can easily
be
derived from the crystal symmetry by removing the
translational part of every symmetry operator and adding a center of
symmetry (if it did not exist previously). For example, if the symmetry
operations of a crystal are those of the P21 space
group, the symmetry operators of the
corresponding Patterson Function will be:
P21
(x, y,
z) (-x, 1/2+y, -z)
Patterson (x,
y, z) (-x, y, -z) (-x, -y,
-z) (x, -y, z)

 The impossibility
of measuring the relative phases
among the diffracted beams, Φ(hkl), makes unfeasible a direct calculation of
the
electron density function
(Formula 1, below), which would provide us the atomic positions within
the unit cell. This was remedied only after 1934
when Arthur
Lindo Patterson
(1902-1966) introduced his brilliant idea, thereby obtaining the first solution
to the phase problem..., as he demonstrated in
his article entitled A
Fourier Series Method for the Determination of the Components of
Interatomic Distances in Crystals, A.L. Patterson (1934) Phys. Rev.,
46, 372-376. If you
have no access to this article in Phys.Rev., you may get a
photograph of its pages (except the last one), as provided by
the University
of Illinois.
The impossibility
of measuring the relative phases
among the diffracted beams, Φ(hkl), makes unfeasible a direct calculation of
the
electron density function
(Formula 1, below), which would provide us the atomic positions within
the unit cell. This was remedied only after 1934
when Arthur
Lindo Patterson
(1902-1966) introduced his brilliant idea, thereby obtaining the first solution
to the phase problem..., as he demonstrated in
his article entitled A
Fourier Series Method for the Determination of the Components of
Interatomic Distances in Crystals, A.L. Patterson (1934) Phys. Rev.,
46, 372-376. If you
have no access to this article in Phys.Rev., you may get a
photograph of its pages (except the last one), as provided by
the University
of Illinois.