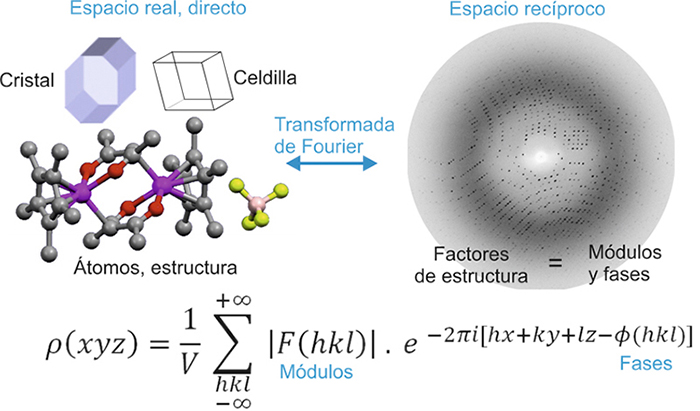
Aspecto de algunas de las salas del
primer computador, conocido con el nombre de ENIAC (Electronic
Numerical Integrator and Computer, 1945).
Aspecto de algunas de las salas del
primer computador, conocido con el nombre de ENIAC (Electronic
Numerical Integrator and Computer, 1945).
El primer computador, ENIAC, se
construyó por encargo del ejército de los Estados
Unidos en 1943 para
realizar cálculos balísticos. Reemplazaba a 200
personas encargadas de
calcular las tablas de tiro. Estaba compuesto por 70.000
resistencias, 10.000 condensadores, 1.500 relés,
6.000 conmutadores
manuales y 17.486 válvulas, que debido a su fragilidad
debían cambiarse
frecuentemente (fueron necesarios 19.000 cambios en los nueve
años en los que esta máquina
estuvo en servicio).