
 La
imposibilidad de medir las fases, Φ(hkl),
relativas entre los haces dispersados en un experimento de
difracción hace inviable el cálculo directo de la
función de densidad electrónica
(ver Fórmula 1, más abajo), cuyos
máximos nos
proporcionarían las posiciones de los átomos en
la celdilla elemental. Fue sólo a partir de 1934,
cuando Arthur
Lindo Patterson (1902-1966) introdujo
su
brillante idea,
obteniéndose así la
primera solución
al problema de las fases...,
tal como demostró en su
brillante artículo titulado A
Fourier Series Method for the Determination of the Components of
Interatomic Distances in Crystals, A.L. Patterson (1934) Phys. Rev.,
46, 372-376. En caso de no poder acceder al
artículo de Phys.Rev., puede Vd. disponer de una
fotografía de sus páginas (excepto la
última), tal como proporciona la Universidad
de Illinois.
La
imposibilidad de medir las fases, Φ(hkl),
relativas entre los haces dispersados en un experimento de
difracción hace inviable el cálculo directo de la
función de densidad electrónica
(ver Fórmula 1, más abajo), cuyos
máximos nos
proporcionarían las posiciones de los átomos en
la celdilla elemental. Fue sólo a partir de 1934,
cuando Arthur
Lindo Patterson (1902-1966) introdujo
su
brillante idea,
obteniéndose así la
primera solución
al problema de las fases...,
tal como demostró en su
brillante artículo titulado A
Fourier Series Method for the Determination of the Components of
Interatomic Distances in Crystals, A.L. Patterson (1934) Phys. Rev.,
46, 372-376. En caso de no poder acceder al
artículo de Phys.Rev., puede Vd. disponer de una
fotografía de sus páginas (excepto la
última), tal como proporciona la Universidad
de Illinois.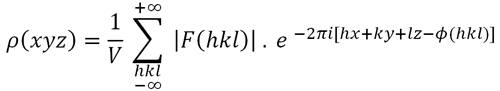
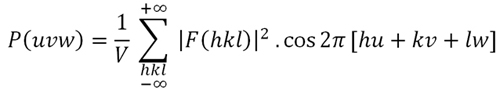
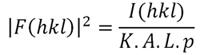

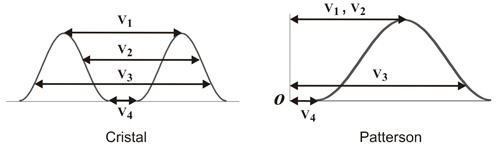
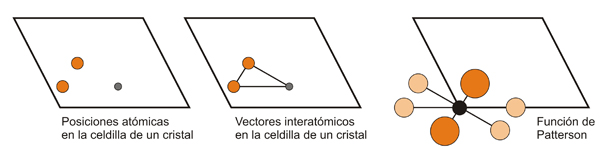
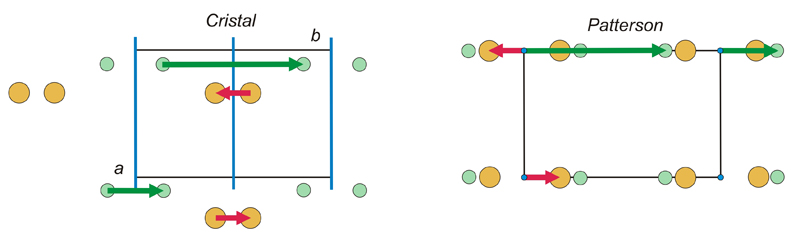
 Resolver
una función de Patterson implica deducir las coordenadas de
los
átomos en el cristal (normalmente las de los
átomos con mayor número de
electrones) a partir de las coordenadas de los máximos del
mapa de
Patterson, y puede entenderse que en general no es una tarea sencilla.
Al menos no lo era hasta que en 1935 David
Harker (1906-1991), un "aprendiz de
cristalógrafo", se diera
cuenta de una circunstancia especial que facilitaba
significativamente
la interpretación de la función y de la que Arthur
L. Patterson no había sido consciente.
Resolver
una función de Patterson implica deducir las coordenadas de
los
átomos en el cristal (normalmente las de los
átomos con mayor número de
electrones) a partir de las coordenadas de los máximos del
mapa de
Patterson, y puede entenderse que en general no es una tarea sencilla.
Al menos no lo era hasta que en 1935 David
Harker (1906-1991), un "aprendiz de
cristalógrafo", se diera
cuenta de una circunstancia especial que facilitaba
significativamente
la interpretación de la función y de la que Arthur
L. Patterson no había sido consciente.| Grupo Espacial | Posiciones equivalentes en el cristal |
Vectores de Patterson (diferencia de coordenadas en el cristal) |
| Pm |
(x, y, z) (x, -y, z) | <0, 2y, 0> línea Harker |
| P21 | (x, y, z) (-x, 1/2+y, -z) | <2x, 1/2, 2z> plano Harker |
| Número | u | v | w | Valor relativo de la función de Patterson |
| 1 | 0 | 0 | 0 | 999 |
| 2 | 0.50 | 0.50 | 0.45 | 342 |
| 3 | 0 | 0.05 | 0.50 | 337 |
| 4 | 0.51 | 0.45 | 0.95 | 137 |
| 5 | 0.26 | 0.92 | 0.14 | 129 |
| Simetría del Grupo Espacial P21/c | |||
| (x, y, z) | (-x, 1/2+y, 1/2-z) | (-x, -y, -z) | (x, 1/2-y, 1/2+z) |
| Máximos Harker | (-x, 1/2+y, 1/2-z) | (-x, -y, -z) | (x, 1/2-y, 1/2+z) |
| (x, y, z) | <2x, -1/2, -1/2+2z> | <2x, 2y, 2z> | <0, -1/2+2y, -1/2> |
| Máximos Harker | <2x, 1/2, 1/2+2z> | <2x, 2y, 2z> | <0, -1/2+2y, 1/2> |
| Simetría del mapa de Patterson | |
| (x, y, z) |
(-x, y, -z) |
| (-x, -y, -z) | (x, -y, z) |